روشهای دسلولار کردن قلب جنین و بررسی سلول دار شدن اسلایسهای بافت قلب در رت
بیماریهای قلبی عروقی بهعنوان عامل اصلی مرگ و میر جهانی اهمیت اجرای طرح را افزایش دادهاند. پزشکی بازساختی با فناوریهای پیشرفتهتر نسبت به درمانهای موقت، این چالش را بهبود میبخشد. استفاده از ویژگیهای خاص ECM در مهندسی بافت و پزشکی بازساختی با هدف بازگرداندن عملکرد بافتهای آسیبدیده کاربرد دارد. این مطالعه بر بررسی سلولزدایی از بافت سالم جنین و استفاده از آن در بازسازی حفره صفاقی رتها با استفاده از اسکافولد و ECM متمرکز است.
بررسی تظاهرات قلبی در انسفالوپاتی های صرعی تکاملی DEE مرتبط با جهش در کانال های یونی
تاخیر تکاملی و ناتوانی ذهنی به عنوان انسفالوپاتیهای تکاملی شناسایی میشوند که ممکن است در اوایل پس از تولد ظاهر شوند و بیش از یک پنجم این بیماران به نوعی از اپیلپسی مبتلا هستند. در مواردی که انسفالوپاتی تکاملی با صرع همراه است، تشنجهای مکرر میتواند ناتوانی ذهنی را تشدید کند و منجر به انسفالوپاتیهای صرعی-تکاملی شود. بررسیهای ژنتیکی نشان میدهد جهشهای ژنتیکی در این بیماران شایع است که میتواند پاتوفیزیولوژی این اختلالات باشد. شناخت این جهشها میتواند به درمانهای اختصاصی منجر شود. ارتباطات قلبی-عصبی نیز شناخته شده است و بیماران مبتلا به این سندرمها ممکن است مشکلات قلبی داشته باشند. هدف مطالعه بررسی تظاهرات قلبی در کودکان با سندرمهای صرعی تکاملی و پیشگیری از SUDEP و اختلالات قلبی است.
اثربخشی بتا بلاکرها بر خونریزی واریس در کودکان مبتلا به فشار خون پورتال
خونریزی واریس ناشی از فشار خون پورتال (PHT) شایعترین علت خونریزی دستگاه گوارش فوقانی در کودکان است. مسدودکنندههای بتا به عنوان داروی درمانی برای کاهش فشار ورید پورتال (PVP) استفاده میشوند، اما دادهها نتایج متناقضی در پیشگیری از خونریزیهای پرفشاری خون پورتال در کودکان نشان دادهاند. هدف این طرح ارزیابی اثربخشی مسدودکنندههای بتا در پیشگیری از خونریزی واریس در کودکان مبتلا به PHT و بررسی برتری بتا بلوکرهای جدیدتر نسبت به بلوکرهای معمولی است.
بررسی ارگانیک اسیدها در مایع آمونیوتیک مبتلایان به ناهنجاریهای جنینی به روش GC/MASS
طرح "بررسی ارگانیک اسیدها در مایع آمونیوتیک مبتلایان به ناهنجاریهای جنینی به روش GC/MS" به دلیل امکان شناسایی زودهنگام ناهنجاریها و کاهش خطرات و هزینههای روشهای تهاجمی، اهمیت ویژهای دارد. این طرح با استفاده از روشهای غیرتهاجمی، به بهبود کیفیت مراقبتهای پیش از تولد و ارتقای دانش متابولومیکس کمک میکند. اطلاعات حاصل میتواند به پزشکان و والدین در مدیریت بارداری و تصمیمگیریهای پس از تولد یاری رساند.
بررسی نتایج جراحی بیماران جابجایی عروق بزرگ عمل شده در مرکز طبی کودکان از سال 1392 تا 1403
بیماری TGA یک ناهنجاری قلبی-عروقی پیچیده است که نیاز به جراحی اصلاحی زودهنگام دارد. ارزیابی عملکرد دریچههای قلبی با اکوکاردیوگرافی قبل و بعد از جراحی، به شناسایی مشکلات و تنظیم درمان کمک میکند و میتواند به کاهش عوارض، بهبود پروگنوز، پیشگیری از مشکلات طولانیمدت و توسعه دانش پزشکی منجر شود. اجرای این طرح در مرکز طبی اطفال منجر به بهبود روشهای درمانی و کاهش مورتالیتی در بیماران TGA خواهد شد.
ارزیابی نتایج جراحی بیماران با اختلال منشاگیری عروق کرونری از شریان ریوی عمل شده در مرکز طبی کودکان از سال 1392 تا 1403
متن به بررسی ضرورت اجرای طرحی برای بهبود جراحی بیماران مبتلا به سندرم anomalous left coronary artery from the pulmonary artery (ALCAPA) میپردازد. این سندرم نادر، یکی از علل ایسکمی میوکارد در کودکان است و انتخاب روش جراحی مناسب برای کاهش مورتالیتی و عوارض پس از جراحی اهمیت زیادی دارد. استفاده از مودالیتههای تشخیصی مانند اکوکاردیوگرافی قبل از جراحی و بررسی دقیق فرآیندهای حین جراحی میتواند در بهبود نتایج و کاهش هزینههای نظام سلامت مؤثر باشد. هدف اصلی این مطالعه، بررسی ارتباط بین اکوکاردیوگرافی و پیشآگهی جراحی در بیماران ALCAPA است تا از عوارض جراحی و مورتالیتی پیشگیری شود.
بررسی عملکرد قلب به روش spackle tracking echocardiography برای پیشگیری از عوارض myocardial iron overload در بیماران مبتلا به تالاسمی ماژور در مرکز طبی کودکان در سال 1403
روش T۲MRI به عنوان استاندارد طلایی برای ارزیابی اضافه بار آهن قلبی در بیماران تالاسمی ماژور شناخته میشود اما به دلیل هزینه بالا و دسترسی محدود، استفاده از روشهای غیرتهاجمی و کمهزینه مانند اکوکاردیوگرافی اهمیت دارد. اکوکاردیوگرافی میتواند در تشخیص زودهنگام مشکلات قلبی موثر باشد و روشهای مختلفی مانند spackle tracking، doppler و tissue doppler imaging در آن به کار میروند. spackle tracking در پیشبینی نارسایی قلبی و تغییرات قلبی مفید است و میتواند در بیماران بتا تالاسمی ماژور بدون علائم قلبی به کار رود. این روش هزینههای کمتری به سیستم سلامت و بیماران تحمیل میکند و تشخیص اختلالات قلبی را تسریع میبخشد. هدف این طرح تحقیقاتی استفاده از این روش برای مشاهده تغییرات هر چهار حفره قلبی و تشخیص زودرس اختلالات قلبی است. ضرورت اجرای طرح کاهش هزینههای نظام سلامت و بیماران است.

ارزیابی مجدد تأثیر واریانت نوین احتمالاً بیماریزا c.1286_1288delAGA در ژن ATP8A2: پیگیری 7 ساله با بینشهای بالینی، ژنتیکی و ACMG در یک خانواده ایرانی
سندرم CAMRQ یک اختلال نادر نورولوژیکی است که با آتاکسی مخچهای، ناتوانی ذهنی و آتروفی مخچه مشخص میشود. این مطالعه فنوتیپ بالینی یک پسر 10 ساله مبتلا به سندرم CAMRQ4 را طی 7 سال پیگیری کرده است. بیمار علائم معمول شامل اختلال هماهنگی حرکتی، اختلال شناختی و اختلالات تعادلی را نشان داد. تجزیهوتحلیل ژنتیکی یک واریانت حذفی درونچارچوبی هموزیگوت (c.1286_1288delAGA) در ژن ATP8A2 را شناسایی کرد که بر اساس تفکیک خانوادگی، بهعنوان واریانت بیماریزا تأیید شد. این یافته طیف جهشی سندرم CAMRQ را گسترش میدهد و بر اهمیت آزمایشهای ژنتیکی جامع در تشخیص اختلالات نورولوژیکی نادر تأکید میکند. تحقیقات بیشتر برای بهبود راهبردهای مراقبت و مدیریت این بیماران ضروری است.

واریانت نوین EDARADD در دیسپلازی اکتودرمال کشفشده توسط توالییابی تمام اگزوم
دیسپلازی اکتودرمال (ED) گروهی از اختلالات ژنتیکی است که بر ساختارهای مشتق از اکتودرم مانند دندانها، مو، ناخنها و غدد عرق تأثیر میگذارد. این مطالعه یک دختر 20 ماهه ایرانی را با تظاهرات بالینی دیسپلازی اکتودرمال هیپوهیدروتیک (HED) شامل ناهنجاریهای دندانی شدید، موهای کمپشت و انیکودیستروفی بررسی کرده است. توالییابی تمام اگزوم (WES) یک واریانت نوین تغییر چارچوب خواندن (c.36dupT) در ژن EDARADD را شناسایی کرد که بر اساس معیارهای ACMG بهعنوان بیماریزا طبقهبندی شد. تجزیهوتحلیل تفکیک الگوی وراثت اتوزومال مغلوب را تأیید کرد و هر دو والد بهعنوان ناقلان هتروزیگوت شناسایی شدند. این یافته بینشهای مهمی درباره پایه ژنتیکی دیسپلازی اکتودرمال ارائه میدهد و بر اهمیت ادغام دادههای بالینی با آزمایشهای ژنتیکی پیشرفته برای تشخیص دقیق و درمان شخصیسازیشده بیماران مبتلا به اختلالات ژنتیکی نادر تأکید میکند.
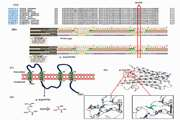
شناسایی و مشخصهیابی عملکردی دو واریانت نوین SRD5A2 در خواهران ایرانی مبتلا به نقص 5α-ردوکتاز نوع 2: گسترش طیف جهشی و پیامدها برای تشخیص ژنتیکی
اختلالات تمایز جنسی (DSD) شرایط مادرزادی هستند که رشد طبیعی بافتهای جنسی را به دلیل ناهنجاریهای ژنتیکی مختل میکنند. نقص 5α-ردوکتاز نوع 2 ناشی از جهشهای ژن SRD5A2، تبدیل تستوسترون به دیهیدروتستوسترون (DHT) را که برای تمایز اندام تناسلی مردانه ضروری است، مختل میکند. این مطالعه دو خواهر از یک خانواده ایرانی با ازدواج فامیلی را بررسی کرده که هر دو با DSD نوع 46,XY مراجعه کردند. ارزیابیهای بالینی و بیوشیمیایی کاریوتایپ 46,XY و نسبتهای تستوسترون به DHT افزایشیافته را نشان دادند. آزمایش ژنتیکی مولکولی دو واریانت نوین هموزیگوت در ژن SRD5A2 را شناسایی کرد: c.314G>C, p.(Arg105Thr) و c.445+5G>C. تجزیهوتحلیل تفکیک نشان داد والدین ناقلان هتروزیگوت هستند. تحلیلهای بیوانفورماتیک نشان دادند واریانت p.(Arg105Thr) ساختار پروتئین را بیثبات میکند و واریانت c.445+5G>C احتمالاً برش و وصل طبیعی را مختل میکند. این یافتهها طیف جهشی SRD5A2 را گسترش میدهند و بر اهمیت تشخیص ژنتیکی زودهنگام در DSD، بهویژه در جمعیتهای با نرخ بالای ازدواج فامیلی، تأکید میکنند.